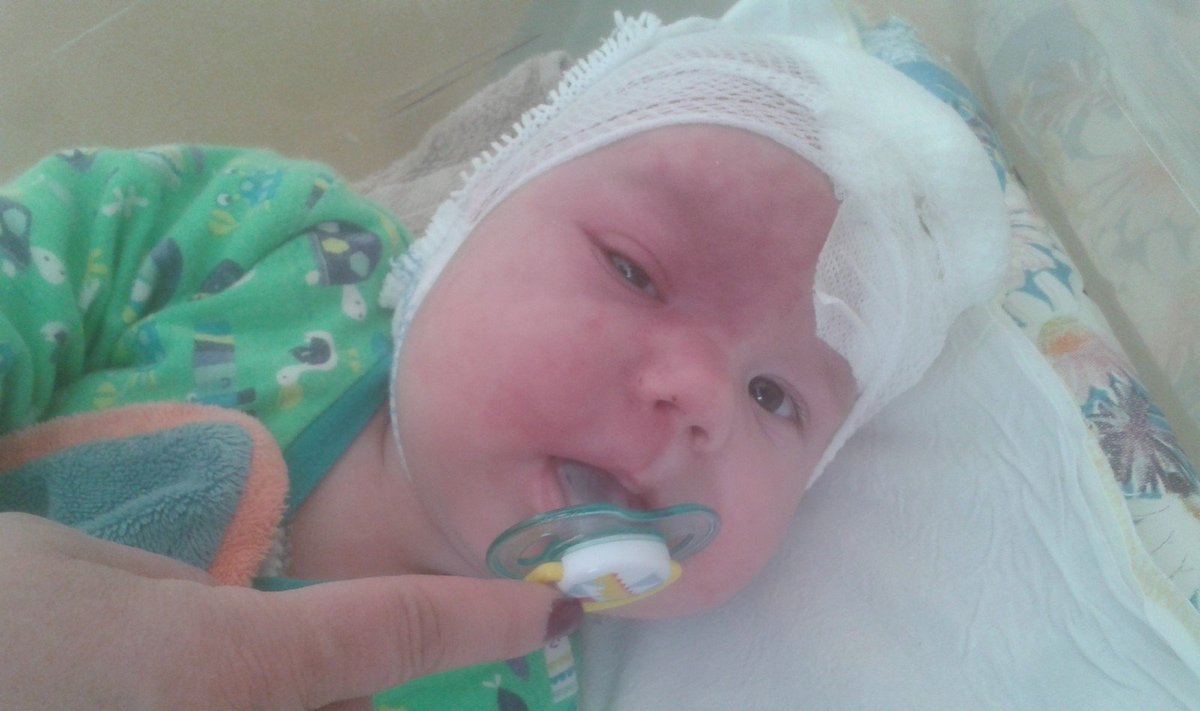
„Sūnus gimė su dėme, kitų vadinama vyndėme – ji raudona, dengė pusę veido. Gydytojai sakė, kad tai galėtų būti šis sindromas, bet jis labai retai - tik 15 proc. atvejų - išsiduoda vizualiai“, - pasakojimą pradeda moteris.
Kūdikiui vasarį duotas siuntimas į klinikas Vilniuje, kur dėmė buvo pašalinta lazeriu. Operacija buvo kompensuota, nes dėmės nepašalinus gali grėsti sudėtingos komplikacijos.
„Viskas buvo gerai, tada staiga, prieš porą dienų iš niekur nieko prasidėjo vienos kūno pusės traukuliai. Vaikas pavalgė ir tiesiog pusė kūno ėmė drebėti. Iškvietėme greitąją ir išvykome į ligoninę Šiauliuose. Ten ėmė spėlioti, kad kraujas išsiliejęs į smegenis, nes esą buvo smurtauta prieš vaiką. Tada nuvykome į Kauno klinikas, kur patvirtino kraujo išsiliejimą ir diagnozavo tikrąją to priežastį – Sturge Weber sindromą“, - pasakojo K. Tiškuvienė.
Šiuo metu dar per anksti kalbėti, kaip liga paveiks vaiko ateitį.
„Daug kas neaišku, nes sindromas gali pasireikšti labai įvairiai. Kadangi sindromas toks retas, sunku spėlioti, kas bus, o kas ne. Vaikas gali būti autistas, tai priklauso nuo traukulių gausumo. Traukulius stabdo vaistais, tačiau vakar naktį jie vėl buvo priepuolis. Dar reikės atlikti daug tyrimų, kad būtų aiškiau“, - kalbėjo pašnekovė.
Dar viena grėsmė – glaukoma.
„Akis tokia tarsi „išėjusi į priekį“, atrodo it sutinęs vokas, nes vidinės kraujagyslės išstorėjusios. Sindromas gali sukeltį glaukomą. Gydytojai rytoj tirs akį, akispūdį, bet sakė, kad jau mato kažką negero vienoje akyje“, - prastomis žiniomis dalijosi moteris.
Moteris šiuo metu prašo visuomenės pagalbos, nes po skyrybų neturi finansinių galimybių rūpintis sūnumi.
„Jei aš nebūsiu stipri, kas kitas vaiku pasirūpins? Su vyru išsiskyrėme dar prieš vaiko ligą. Jis smurtavo, gėrė, neparnešdavo algos. Penktadienį prieš įvykstant vaiko priepuoliui, man nutraukė pašalpas – kadangi išsiskyriau, man pirmiausia, sako, reikia prisiteisti alimentus. Dabar esu visai be pajamų. Kol susitvarkysiu visus teismo reikalus, praeis mažiausiai porą mėnesių, o dabar dar viskas nusikėlė, nes su vaiku visą laiką esu ligoninėje. Bet aš netikiu, kad ir po teismo prisiteisiu pinigų, nes buvęs vyras neturės iš kur jų mokėti“, - sakė K. Tiškuvienė.
Moteris pasakojo, kad susirgus vaikui ji su mama kreipėsi į savo buvusį vyrą prašydama sauskelnių, mišinukų, tačiau sulaukė atžagaraus atsakymo. Su Kristina jis susisiekė tik dabar, feisbuke išplitus moters prašymui padėti. „Gal gėda jau jam, bet tikrai ne sąžinė atsibudo“, - mano pašnekovė.
Gydymas, žinoma, yra nemokamas, tačiau kainuoja pragyvenimas, sauskelnės. Galintys padėti daugiau informacijos, kaip tai padaryti, ras čia (arba rašykite adresu sveikata@delfi.lt ir atsiųsime detales).
Liga nustatoma kliniškai
Apie šį retą sindromą DELFI komentuoja Milda Endzinienė, Lietuvos sveikatos mokslų universiteto Neurologijos klinikos profesorė Kauno klinikų Fakomatozių centro vadovė, Retų ir nediagnozuotų ligų koordinacinio centro vadovė ir Milda Dambrauskienė, Kauno klinikų vaikų neurologė, Retų ir nediagnozuotų ligų koordinacinio centro koordinatorė.
- Prašau papasakokite apie šį sindromą? Kiek jis retas? Ar būta pacientų Kauno klinikose?
- Sturge-Weber sindromas priklauso fakomatozių, tai yra centrinę nervų sistemą ir odą pažeidžiančių ligų, grupei. Retų ligų informacijos portalo ORPHANET duomenimis, Sturge-Weber sindromas nustatomas vienam iš 20 000–50 000 naujagimių, tad Lietuvoje toks vaikas galėtų gimti kartą per metus arba dvejus. Tai yra retas įgimtas sindromas, kuris pažeidžia odą, galvos smegenis ir akis. Kitos dažniau pasitaikančios fakomatozių grupės ligos – I ir II tipo neurofibromatozė, tuberozinė sklerozė ir Von Hippel-Lindau liga.
Sturge-Weber sindromas nėra paveldimas. Jį nulemia mutacija GNAQ gene, kuri atsiranda atsitiktinai ir dėl nežinomų priežasčių. Kai kurie autoriai išskiria tris sindromo tipus: I tipui būdingi odos pakitimai ir neurologiniai simptomai. Antram tipui pasireiškia odos požymiai ir glaukoma, tačiau nepažeista nervų sistema. Trečiajam tipui būdingas izoliuotas nervų sistemos pažeidimas, be odos požymių ir glaukomos.
Šiuo metu Kauno klinikų Fakomatozių centro, kuriam vadovauja prof. Milda Endzinienė, specialistai prižiūri penkis tokius pacientus, kuriems dėl sindromo pasireiškia priepuoliai, akių problemos. Kadangi liga pažeidžia ne vieną organizmo sistemą, į tokių pacientų priežiūrą ir gydymą įtraukiama visa komanda: neurologas, oftalmologas, neurochirurgas, radiologas, dermatologas, psichologas, kineziterapeutas ir kiti specialistai.
- Kaip liga nustatoma? Ar ant mažojo paciento veido buvusi raudona dėmė susijusi?
- Sturge-Weber sindromas nustatomas kliniškai, tai yra, apžiūrint pacientą, nes jo klinikiniai požymiai yra pakankamai akivaizdūs: pirmiausia pastebimos kapiliarų malformacijos veide, vėliau gali paaiškėti, kad panašių kraujagyslių apsigimimų toje pačioje pusėje galvos smegenyse ir akyje. Tipiškas ligos požymis yra jau gimimo metu matoma „vyno“ arba „portveino dėmė“, dar vadinama „ugniniu apgamu“, kuris apima vieną veido pusę arba abi, kartais tik kaktą ir akį, kartais – ir apatinę veido dalį, galinti išplisti į kaklą, ranką ar liemenį.
Galvos smegenyse būna minkštųjų dangalų angiomos, smegenų dalies atrofija. Pažeidimo apimtį patikslinti padeda galvos smegenų kompiuterinės tomografijos ar magnetinio rezonanso tomografijos tyrimas. Keturiems iš dešimties pacientų nustatomas intelekto sutrikimas. Trys ketvirtadaliai jų – patiriantys epilepsijos priepuolius.
Vienos kūno pusės galūnių nusilpimas, nejudrumas (hemiplegija) priešingoje pusėje nei veido angioma būna kas trečiam ligoniui. Kartais toks nusilpimas atsiranda po pirmojo epilepsijos priepuolio ir ryškėja jiems kartojantis. Pastaruoju metu išsamiau tiriami endokrininės sistemos sutrikimai, būdingi šiems ligoniams (augimo sutrikimas, skydliaukės hormonų stoka).
- Ar sindromą lengva supainioti su kita liga?
- Dažniausiai kūdikiai turi hemangiomų, kurios išryškėja pirmaisiais mėnesiais, bet nebūtinai matomos iškart po gimimo. Hemangiomoms būdinga tai, kad antraisiais gyvenimo metais jos neretai išnyksta savaime. „Vyno dėmės“ savaime neišnyksta, joms reikalingas gydymas lazeriu ir paprastai prireikia ne vienos procedūros. Kita vertus, tai nėra nuosprendis, nes iš visų pacientų, turinčių „vyndėmę“, tik 8–20% pasireiškia neurologinių simptomų. Dar rečiau pasitaiko atvirkštinė situacija – kai pakitimų odoje neturinčiam pacientui nustatomi neurologiniai šio sindromo požymiai.
- Kokios šio sindromo gydymo galimybės?
- Sturge-Weber sindromo atveju pagrindinis gydymo tikslas – epilepsijos priepuolių kontrolė, galvos skausmų ir į insultą panašių epizodų, taip pat glaukomos gydymas.
Vaikams, kuriems nustatytas Sturge-Weber sindromas, dažnai pasitaiko insultą primenantys epizodai, kuriems būdingi laikini neurologiniai simptomai, praeinantys per kelias dienas ar savaites. Taip pat šiems ligoniams gali kartotis migrenos priepuoliai, kuriuos veiksmingai galima gydyti specifiniais vaistais.
Epilepsijos priepuoliai pasireiškia iki 75–90 proc. pacientų, kuriems nustatytas šis retas sindromas. Paprastai priepuoliai prasideda pirmaisiais gyvenimo mėnesiais, jie gali būti labai dažni. Kai kuriems pacientams priepuolius puikiai pavyksta sustabdyti vos paskyrus vaistus nuo epilepsijos, tačiau daliai pacientų priepuoliai linkę kartotis ir taip sutrikdyti pažintines funkcijas.
Norint jas išsaugoti, epilepsijos gydymas skiriamas iškart po pirmojo priepuolio arba netgi dar anksčiau, kol priepuolių nėra buvę. Jei priepuolių nepavyksta sustabdyti netgi skiriant du ar tris vaistus nuo epilepsijos, tuomet reikėtų apsvarstyti kitas gydymo galimybes – ketogeninę dietą arba epilepsijos chirurgiją. Tebevyksta diskusijos, kada reikėtų įsikišti neurochirurgams ir taikyti chirurginį epilepsijos gydymą. Visais atvejais dėl operacijos apimties nusprendžiama įvertinus ligonio būklę, pasikartojančius priepuolius ir numatomas chirurginio gydymo pasekmes. Tačiau reikia nepamiršti, kad sustabdžius atkaklius priepuolius galima užtikrinti geresnę vaiko raidą ar išsaugoti intelektinius gebėjimus.
Visame pasaulyje pacientai, kuriems diagnozuotas Sturge-Weber sindromas, buriasi į organizacijas, kurios remia naujų diagnostikos ir gydymo būdų paieškas, aktyviai dalijasi informacija ir padeda šeimoms, kurioms tenka išgirsti tokią diagnozę. Lietuvoje veikianti Vaikų retų ligų asociacija taip pat prisideda prie šios veiklos, vienija ir fakomatozėmis sergančius asmenis. Universitetinės ligoninės, bendradarbiaudamos su pacientų organizacijomis, gali pasiūlyti visapusišką sveikatos priežiūrą ir psichologinę pagalbą.
